
Regulation of nephron transport, salt-sensitive hypertension and cross-talk between tubules and vessels
We study regulation of salt and water absorption along the renal nephron, how these processes alter the renal vasculature and how disruptions in this regulation can lead to hypertension, a disease afflicting 25-30% of the U.S. adult population. Many of the hormones and factors that regulate systemic vascular tone also regulate solute and solvent absorption along the nephron including angiotensin II, endothelin and luminal flow. The reactive oxygen species nitric oxide (NO) and superoxide (O2-) are intermediate signaling molecules for many of the final effects of these hormones and factors. The renal proximal tubules reabsorb about 65% of the filtered Na and water via luminal Na/H exchange, and other Na-coupled transporters. The thick ascending limb of the loop of Henle reabsorbs about 30% of the NaCl filtered by the glomerulus. NaCl enters thick ascending limb cells via the luminal Na/K/2 Cl cotransporter type 2 (NKCC2). Transport defects in both segments have been linked to hypo- and hypertension in animal models and people. Dietary factors such as the amount of fructose and salt one consumes both directly and indirectly alters these processes. Currently we have projects studying how dietary salt and/or sugar alter the actions of angiotensin in proximal tubules, the effects of angiotensin on thick ascending limbs and the balance between flow-stimulated NO and O2- in thick ascending limbs, thereby causing salt-sensitive hypertension.
Regulation of thick ascending limb NaCl absorption by nitric oxide (NO) and superoxide (O2-)
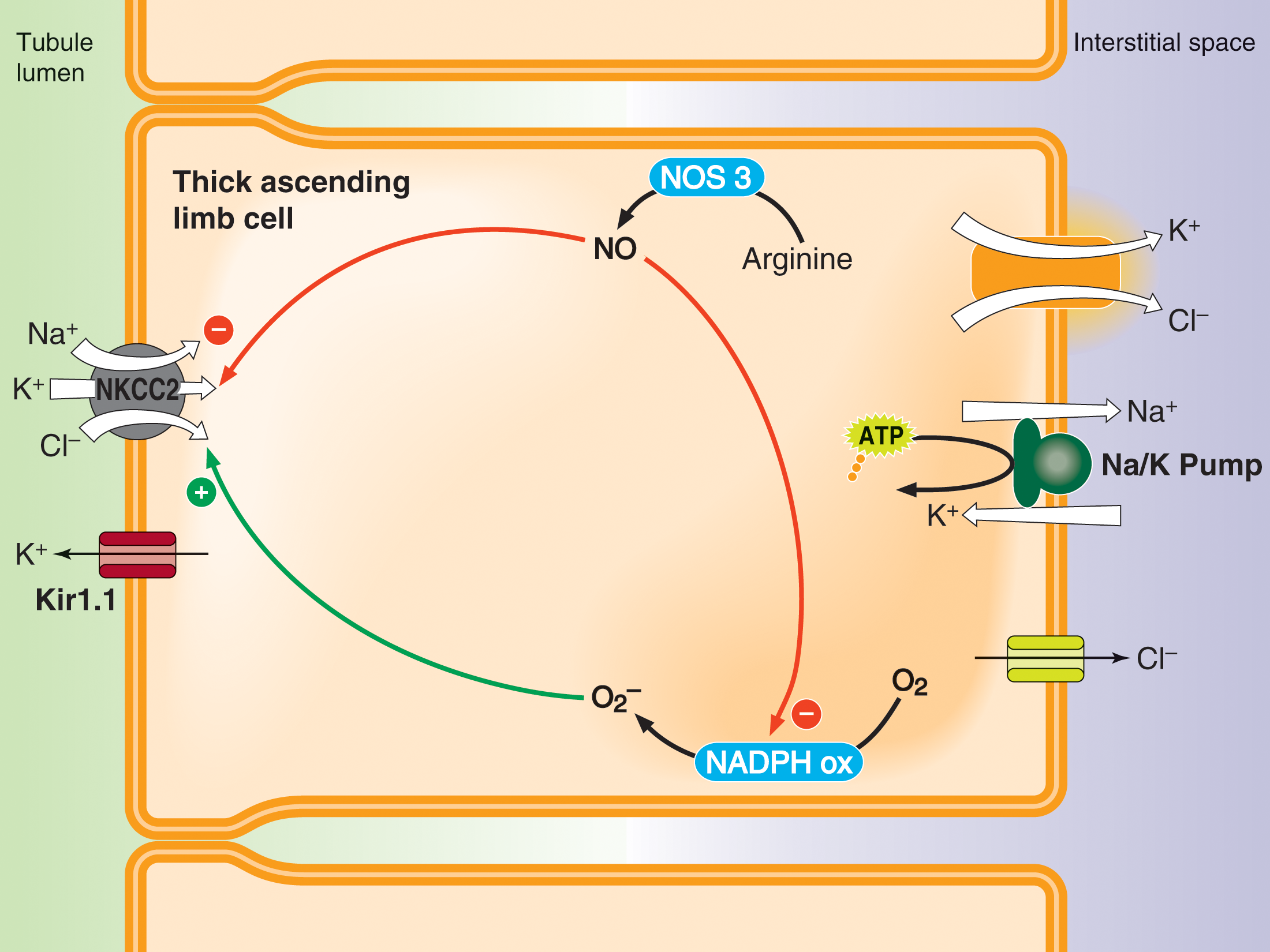
Thick ascending limbs reabsorb about 30% of the NaCl filtered by the glomerulus. NO produced by NO synthase 3 (NOS3) inhibits net NaCl reabsorption by reducing NaCl entry into the cell via NKCC2. Luminal flow and endothelin B, angiotensin type 2 and alpha 2 adrenergic receptor agonists stimulate NOS3 activity. In contrast, O2- enhances net NaCl entry by augmenting NKCC2 activity. O2- is produced by NADPH oxidase, an enzyme composed of several subunits that must assemble to form an active enzyme. Cell stretch caused by luminal flow and angiotensin type 1 receptor agonists activate NADPH oxidase. Flow-induced NO inhibits flow-induced assembly of NADPH oxidase, and thus O2- production. Curiously, angiotensin-induced NO does not have this effect.
Inappropriate thick ascending limb salt reabsorption causes hypertension, the leading cause of “loss of health” world-wide. O2- and NO regulate thick ascending limb NaCl reabsorption, and thus blood pressure. Urine flow causes thick ascending limb cells to stretch and increases the shear on them which stimulates O2- and NO. We reported that flow-stimulated Na reabsorption depends on O2- but that flow-induced NO buffers this by inhibiting NaCl transport and reducing O2-. A high-salt diet increases thick ascending limb flow enhancing NO synthesis via increases in NOS3 expression and activity. Thus, salt-induced increases in luminal flow usually prevent salt retention and increases in blood pressure.
Mechano-transduction of flow-induced stretch and shear occurs via TRPV4 channels and/or cilia with TRPV4-TRPP2 channel activation. We showed that flow-induced stretch stimulates TRPV4 and O2-, and increases intracellular Ca. We also showed that TRPV4 mediates flow-induced increases NO via elevation of intracellular Ca. How cells distinguish between stretch- and shear-induced TRPV4 activation, and the roles of TRPP2 and cilia in flow-induced O2- and NO, and salt-sensitive hypertension are unclear. We hypothesize that in hypertension that thick ascending limbs display abnormally increased TRPV4 activity in response to salt-enhanced luminal flow causing elevated Cai and O2- production. Chronically elevated O2- blunts the ability of flow to stimulate NO. These effects result in salt retention by thick ascending limbs and salt-sensitivity of blood pressure. Thus the ultimate cause of salt-sensitive hypertension is a defect in mechano-transduction.
Click on "Garvin Lab" at the top of the page for more details.
Regulation of thick ascending limb transport by angiotensin II
Hypertension is the leading cause of “loss of health” world-wide. Angiotensin II type 1 receptors (AT1) promote salt retention by stimulating nephron transport, while type 2 receptors (AT2) promote salt excretion. Imbalances favoring the former promote hypertension. Thick ascending limbs reabsorb 25-30% of filtered NaCl through both trans- and paracellular pathways. We and others reported that AT1 activation stimulated protein kinase C and O2-, both of which usually enhance thick ascending limb NaCl absorption. We also showed that stimulating AT2 enhanced NO production. NO inhibits NaCl absorption by activating phosphodiesterase II, which degrades cAMP, and by reducing O2-. However, the role of AT1 in stimulating thick ascending limb NaCl absorption is not well understood, and whether AT2 activation inhibits transport is unknown. Elevated thick ascending limb NaCl absorption and defects in thick ascending limb NO production and signaling contribute to salt-sensitive hypertension and renal injury, but we do not know whether thick ascending limb AT2-induced NO signaling is defective as well. We hypothesize that normally the stimulatory effects of AT1 activation on thick ascending limb NaCl absorption are blunted by AT2-induced NO which reduces cAMP and O2-. Defects in thick ascending limb AT2 signaling promote increases in blood pressure and salt-sensitive hypertension. Understanding how angiotensin II regulates thick ascending limb NaCl absorption will help form an integrated model that explains how angiotensin II regulates urinary Na excretion and thus blood pressure.
Click on "Garvin Lab" at the top of the page for more details.
Hypertension is the leading cause of “loss of health” worldwide. Angiotensin II regulates blood pressure primarily through actions on the kidney, including the proximal tubule. Angiotensin II stimulates transport via activation of angiotensin II type 1 (AT1) receptors and protein kinase C (PKC). When animals are on normal salt angiotensin II nearly maximally stimulate proximal tubule Na reabsorption. Usually when dietary salt is elevated, angiotensin II levels fall by ~70-80%, urinary Na excretion (UNaV) increases and salt is eliminated. More than 15 million Americans consume ≤20% of their calories as fructose. Fructose causes hypertension and cardiovascular disease. We reported that a 20% fructose diet causes salt-sensitive hypertension whereas a 20% glucose+high-salt diet does not. AT1 receptor antagonists prevent fructose-induced hypertension; however, the proximal tubule’s role in fructose-induced salt-sensitive hypertension is unknown.
Our data show that fructose activates either PKC in proximal tubules and that a 20% fructose+high-salt diet enables low concentrations of angiotensin II to stimulate proximal tubule transport. Whether the effects of dietary fructose on the proximal tubule contribute to fructose-induced salt-sensitive hypertension is unknown. We hypothesize that a 20% fructose diet blunts salt-induced natriuresis and causes salt-sensitive hypertension by activating PKCα thereby enabling low concentrations of angiotensin II (induced by a high-salt diet) to stimulate proximal tubule Na reabsorption. If this is correct, many with high blood pressure may benefit by reducing fructose intake or taking Angiotensin II AT1 receptor blockers rather than angiotensin converting enzyme inhibitors.
Click on "Garvin Lab" at the top of the page for more details.
